Computing transfer supports via the web interface
Two types of jobs may be submitted to BOOSTER-WEB:
- Complete phylogenetic analysis (datasets of moderate size);
- Bootstrap support computation.
Complete phylogenetic analysis
If you do not have reference and bootstrap tree files, you can submit a multiple alignment to the run input form (Fasta, Phylip or Nexus format, may be gzipped with .gz extension only), in the “input sequence” field.
In that case, you can choose the workflow to run: (1) PhyML-SMS (for small/medium dataset); or (2) FastTree (for larger datasets).
These two workflows are installed and launched on the Institut Pasteur Galaxy server.
They constist of the following steps:
- Tree inference, two possibilities: (a) Model selection + tree inference using Phyml-SMS; (b) Reference + Bootstrap Tree reconstructions using FastTree.
- Bootstrap support computation using BOOSTER.
Bootstrap support computation
If you have reference and bootstrap tree files, you can also submit BOOSTER jobs directly (run page). In that case, two inputs are required:
- A reference tree file in Newick format (may be gzipped);
- A bootstrap tree file containing all the bootstrap trees (may be gzipped with .gz extension only).
Workflow
Please note that if a multiple alignment file is provided, no tree file will be taken into account.
Clicking the “Run” button will launch the selected analysis and redirect you to a results page, with the following steps:
- The analysis will first be pending, waiting for available resources.
- Then, as soon as the analysis is running, you will be redirected to a waiting page.
- Once analysis is done, the results page shows the following panels:
- Information about the run (identifier, start/end time, number of bootstrap trees analyzed, and output message).
- Links to download the results:
- Tree with FBP (classical) supports.
- Tree with TBE (transfer bootstrap) normalized supports (download Newick format or upload to iTOL).
- Tree with branch labels formatted as follows: “Branch ID|Average transfer Distance|Size of the light side” (download Newick format or upload to iTOL).
- Booster log file with 2 parts:
- Instability score of every taxon (2 columns, “Taxon : Transfer Score”).
- Highly transferred taxa per branch (4 columns: Branch Id, Size of the light side, Average distance, and semicolon separated list of highly transferred taxa with their respective instability score).
- Tree visualizer that highlights branches with a support (FBP or TBE) greater than the cutoff given by the slider.
Generating reference and bootstrap trees
If you want to generate reference and bootstrap trees via other means, you may do so using the following commands (example with 100 bootstrap replicates):
PhyML: Input file: alignment, Phylip format
phyml -i align.phy -d nt -b 100 -m GTR -f e -t e -c 6 -a e -s SPR -o tlr # Output Reference tree: align.phy_phyml_tree.txt # Output Bootstrap trees: align.phy_phyml_boot_trees.txtRAxML: standard bootstrap. Input file: alignment, Phylip format
# Infer reference tree raxmlHPC -m GTRGAMMA -p $RANDOM -s align.phy -n REF # Infer bootstrap trees raxmlHPC -m GTRGAMMA -p $RANDOM -b $RANDOM -# 100 -s align.phy -n BOOT # Output Reference tree: RAxML_bestTree.REF # Output Bootstrap trees: RAxML_bootstrap.BOOT
RAxML: rapid bootstrap. Input file: alignment, Phylip format
# Infer reference tree + bootstrap trees raxmlHPC -f a -m GTRGAMMA -c 4 -s align.phy -n align -T 4 -p $RANDOM -x $RANDOM -# 100 # Output Reference tree: RAxML_bestTree.align # Output Bootstrap trees: RAxML_bootstrap.alignFastTree: you will need to generate bootstrap alignments (Phylip format), such as with goalign. Input file: alignment (Phylip or Fasta format)
# Build bootstrap alignments goalign build seqboot -i align.phy -p -n 100 -o boot -S # Infer reference tree FastTree -nt -gtr align.phy > ref.nhx # Infer bootstrap trees cat boot*.ph | FastTree -nt -n 100 -gtr > boot.nhx # Output Reference tree: ref.nhx # Output Bootstrap trees: boot.nhx
IQ-TREE : booster supports and ultrafast bootstrap. Input file: alignment (Phylip format)
# Infer Reference tree + ultrafast bootstrap trees iqtree-omp -wbt -s align.phy -m GTR -bb 100 -nt 5 # Output Reference tree: align.phy.treefile # Output Bootstrap trees: align.phy.ufboot
Example dataset
You can try BOOSTER-WEB with the following trees inferred from the primate nucleotide alignment taken from “The Phylogenetic Handbook”:
If you also want to infer the reference and bootstrap trees:
- Original alignment: .phy
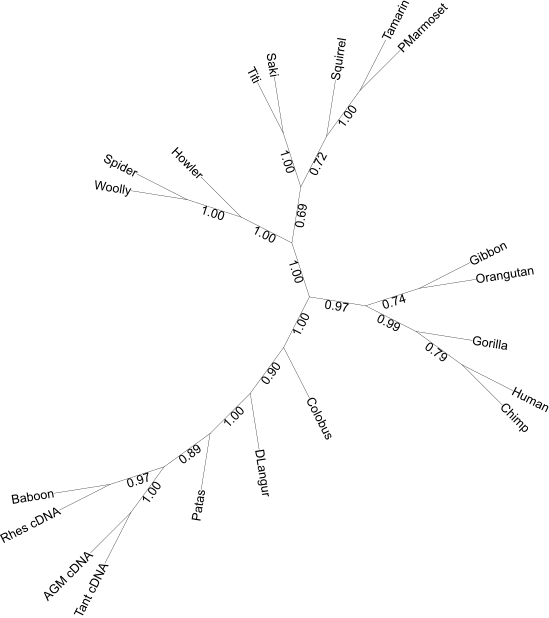
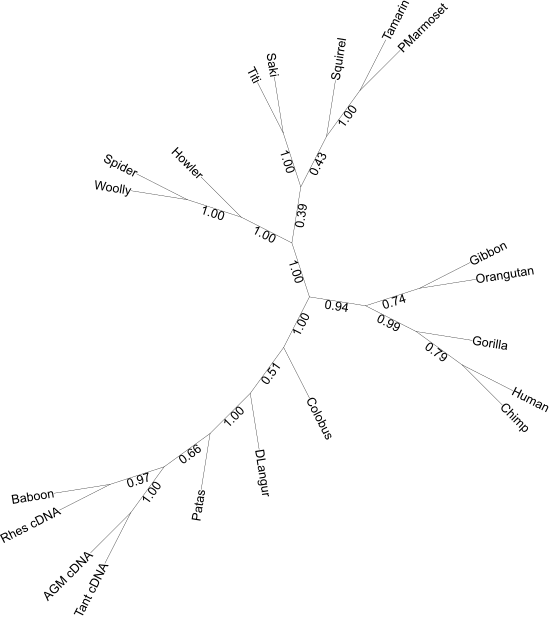
After computingTBE and FBP using BOOSTER-WEB, you should obtain the following trees. TBE supports are larger than FBP supports for all branches. If bootstrap and reference trees were re-computed, some fluctuations of the supports are to be expected.
TBE
FBP
Installing a local version of the web interface
The web interface was developped in Go, and is thus executable on any platform (Linux, MacOS, and Windows). The only thing yo need to do is download the latest release of BOOSTER-WEB from Github, and run it by clicking on the executable.
Then visit the following url: http://localhost:8080.
Computing transfer supports via command line
BOOSTER is also available as a standalone executable (implemented in C). Source and binariy files are available on Github.
Usage: ./booster -i <ref tree file (newick)> -b <bootstrap tree file (newick)> [-d <dist_cutoff> -r <raw distance output tree file> -@ <cpus> -S <stat file> -o <output tree> -v]
Options:
-i : Input tree file
-b : Bootstrap tree file (1 file containing all bootstrap trees)
-a, --algo : bootstrap algorithm, tbe (transfer bootstrap) or fbp (Felsenstein bootstrap) (default tbe)
-o : Output file (optional), default : stdout
-r, --out-raw : Output file (only with tbe, optional) with raw transfer distance as support values in the form of
id|avgdist|depth, default : none
-@ : Number of threads (default 1)
-S : Prints output logs in the given output file (average raw min transfer distance per branches, and average
transfer index per taxa)
-c, --count-per-branch : Prints individual taxa moves for each branches in the log file (only with -S and -a tbe)
-d, --dist-cutoff: Distance cutoff to consider a branch for moving taxa computation (tbe only, default 0.3)
-q, --quiet : Does not print progress messages during analysis
-v : Prints version (optional)
-h : Prints this help
Complete information regarding installation and usage is available on the GitHub page.
Data
The archive containing all data described in the paper can be downloaded from the github page.
Note
PhyML can also directly compute TBE supports (beta). To do this, you will need to download and install PhyML from its github repository:
phyml -i align.phy -d nt -b 100 --tbe -m GTR -f e -t e -c 6 -a e -s SPR -o tlr
# Output tree with supports: align.phy_phyml_tree.txt